

MDD to MDR Gap Analysis — Know Exactly What's Missing Before Your Notified Body Does
Transitioning from the Medical Device Directive to EU MDR 2017/745 is not just an update — it's a complete overhaul of how your Technical Documentation needs to be structured, evidenced, and maintained.
Most companies don't discover their gaps until they're already in front of a Notified Body. That's expensive. That's avoidable.
The MDD to MDR Gap Analysis is a professional Excel tool that maps every critical difference between Directive 93/42/EEC and Regulation (EU) 2017/745 — so your team knows exactly what needs to be done, in what order, and who owns it.
What's inside:
19 documented gaps across 9 regulatory areas — including Clinical Evaluation, Post-Market Surveillance, Technical Documentation, Labelling, UDI, EUDAMED, Software, Risk Management, and Notified Body certification.
Every gap is classified by severity — Major Gap, Minor Gap, or New Requirement — with color coding so you can prioritize at a glance.
Each row includes the exact MDD requirement, the MDR equivalent, and a clear explanation of what changed and why it matters for your submission.
Built-in dropdowns for Status, Priority, and Owner so your team can assign accountability and track progress in real time.
A live Summary Panel that auto-calculates how many gaps are open, closed, and critical — no manual counting needed.
This ready-to-use verification form provides medical device manufacturers with a structured, documented checklist to confirm that UDI labels and UDI carriers meet all requirements of EU MDR (2017/745) and IVDR (2017/746) before a device is placed on the market.
The UDI carrier is the medium on which the UDI is applied or stored in both machine-readable form (AIDC — Automatic Identification and Data Capture) and human-readable form (HRI — Human Readable Interpretation), and must be placed on the label or the device itself, as well as on all higher levels of device packaging.
This form helps your team systematically verify that every label and carrier is complete, correct, and compliant.

Single source of truth for all UDI data your organization must register and maintain. Designed to be filled once per device, reviewed by QA/RA, and used as the data carrier for direct keystroke entry into EUDAMED or for the EUDAMED machine-to-machine (M2M) XML upload.
This Standard Operating Procedure (SOP) provides medical device manufacturers with a clear, step-by-step framework for assigning Unique Device Identifiers (UDIs) and completing device registration in EUDAMED — the European Union's centralized database for medical devices.
The Unique Device Identification (UDI) system enables clear and unambiguous identification of specific medical devices on the market, making traceability easier, improving post-market surveillance, and increasing device safety. As of May 28, 2026, the UDI/Devices module in EUDAMED is mandatory for all manufacturers placing devices on the EU market

FDA Registration Guide for Small Medical Device Businesses
A Practical, Step-by-Step Guide for First-Time FDA Registration
Are you starting a small medical device business and feeling overwhelmed by FDA registration requirements?
This FDA Registration Guide was created specifically for small medical device businesses, startups, and first-time manufacturers who need a clear, practical, and organized way to complete FDA registration and listing — without digging through endless regulations.
This is not legal advice or a generic overview.
It’s a straightforward, actionable guide designed to help you understand what to do, when to do it, and what information you need.
FDA 510(k) Submission Guide — Your Complete Roadmap to FDA Clearance
What's inside — 8 professional sheets:
The Classification and Pathway Guide helps you confirm your device is Class II and choose the right submission route — Traditional, Abbreviated, Special, or De Novo — with review timelines and FDA guidance references for each.
The 510(k) Checklist covers 58 requirements across 11 sections based on 21 CFR 807.87, with dropdowns for Applicable, Status, and Priority — plus color coding that updates automatically as you progress.
The Predicate Device Comparison table walks you through the full substantial equivalence analysis — intended use, technological characteristics, performance, and safety profile — with a built-in SE conclusion template.
The Performance Testing Matrix documents all required tests across five categories: mechanical, electrical safety, biocompatibility, software, and sterilization — with applicable standards, acceptance criteria, and test method fields for every test.
The Submission Dashboard auto-calculates your readiness by section so you always know your percentage complete before submitting to FDA.
The Timeline Planner maps your project across seven phases — from initiation and Q-Sub meeting through FDA review and post-clearance — with realistic duration estimates for every milestone.
And the FDA Glossary covers 26 key terms — 510(k), SE, NSE, IDE, QMSR, GUDID, and more — so your entire team speaks the same language.
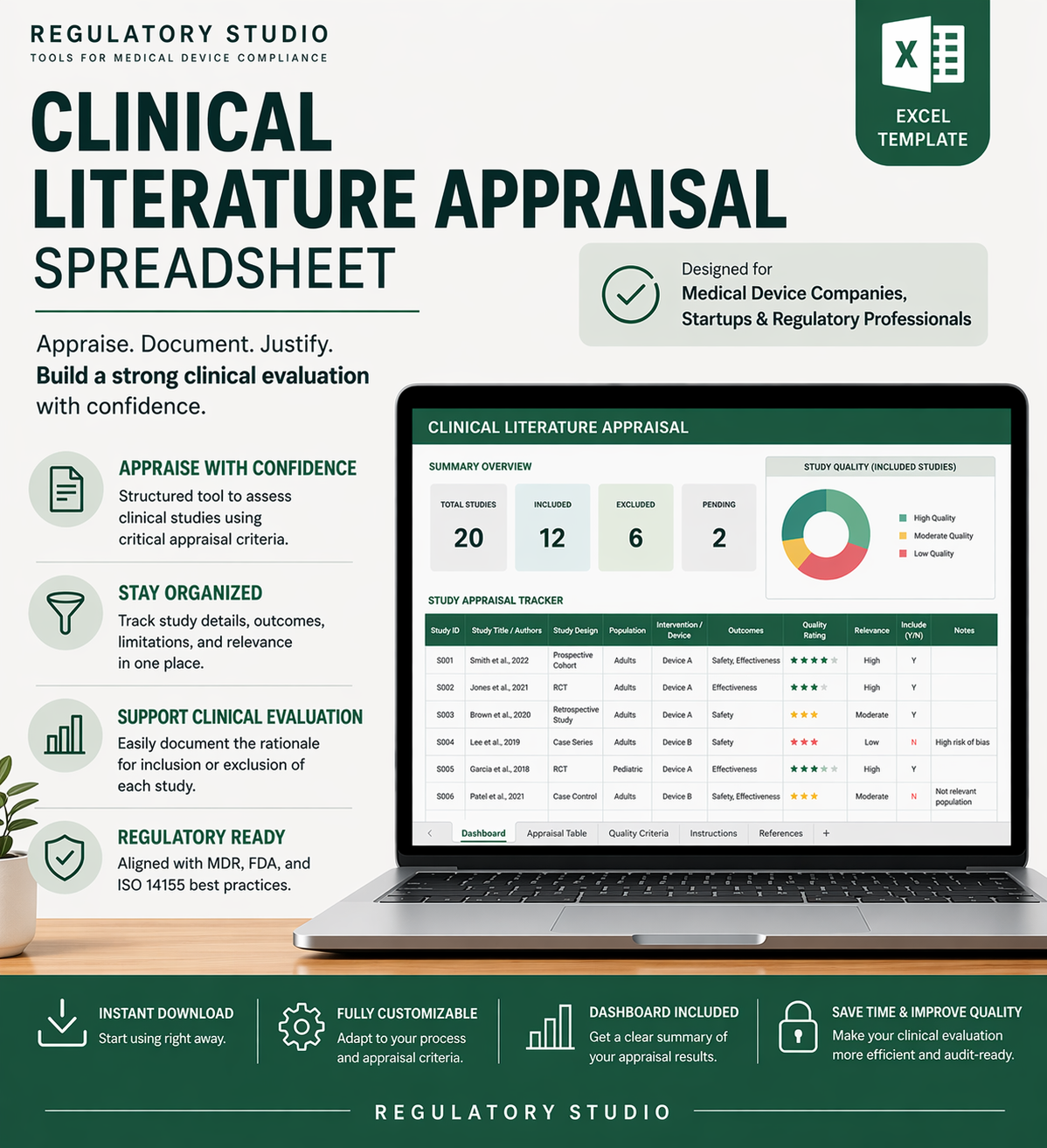
This spreadsheet is designed to organize and simplify your process of appraising clinical literature as part of your post-market surveillance activities or Clinical Evaluation Reports (CERs). It allows you to systematically capture key data points such as study design, patient population, outcomes, and relevance to your device. By structuring and standardizing this information, the spreadsheet helps ensure thorough and consistent evaluations, making it easier to identify trends, gaps, and risks.
No results match your search. Try removing a few filters.